Tutorial: Annotating Genetic Variants with cellink#
This tutorial walks through how to annotate genetic variants within a cellink DonorData (gdata) object using standard tools like VEP, SnpEff, and FAVOR. You will learn how to export variants to VCF, run external annotation tools, integrate the resulting annotations, and visualize variant consequences. These annotations can later be used in downstream analyses such as filtering, visualization, or association testing.
We will use the example dataset onek1k and demonstrate best practices for integrating annotations into the AnnData-like structure gdata.
Setup and Configuration#
We start by importing relevant modules and creating local directories to store input/output files. This ensures that any annotation tools have a consistent file structure to work with.
%load_ext autoreload
%autoreload 2
import cellink as cl
from pathlib import Path
from cellink.resources import get_dummy_onek1k
DATA = Path(cl.__file__).parent.parent.parent / "docs/tutorials/data"
CONFIGS = Path(cl.__file__).parent.parent.parent / "configs"
DATA.mkdir(parents=True, exist_ok=True)
(DATA / "variant_annotation").mkdir(parents=True, exist_ok=True)
variant_file = DATA / "variant_annotation/variants.vcf"
Load Genotype Data (gdata)#
We load the example dataset using get_dummy_onek1k(). (This is a subset of the full OneK1K dataset, which can be downloaded, and prepared using get_onek1k()) The DonorData object contains a .G attribute (gdata) that stores genotype information at the variant level. These variants will be the target of our annotations.
dd = get_dummy_onek1k(config_path="../../src/cellink/resources/config/dummy_onek1k.yaml", verify_checksum=False)
dd
[2025-12-29 01:32:54,052] INFO:root: /Users/larnoldt/cellink_data/dummy_onek1k/dummy_onek1k.dd.h5 already exists
[2025-12-29 01:32:54,052] WARNING:root: No checksum provided, skipping verification
[2025-12-29 01:32:55,305] INFO:root: Loaded dummy OneK1K dataset: (100, 146939, 125366, 34073)
╔═ DonorData(n_donors=100, n_cells_per_donor=[613-2,731], donor_id='donor_id') ═══════════════════════════════╗ ║ ┏━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┳━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┓ ║ ║ ┃ G (donors) ┃ C (cells) ┃ ║ ║ ┡━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━╇━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━┩ ║ ║ │ AnnData object with n_obs × n_vars = 100 × 146,939 │ View of AnnData object with n_obs × n_vars = │ ║ ║ │ │ 125,366 × 34,073 │ ║ ║ │ var: 'chrom', 'pos', 'a0', 'a1', 'AC', │ obs: 'orig.ident', 'nCount_RNA', │ ║ ║ │ 'AC_Hemi', 'AC_Het', 'AC_Hom', 'AF', 'AN', 'ER2', │ 'nFeature_RNA', 'percent.mt', 'donor_id', │ ║ ║ │ 'ExcHet', 'HWE', 'IMPUTED', 'maf', 'NS', 'R2', │ 'pool_number', 'predicted.celltype.l2', │ ║ ║ │ 'TYPED', 'TYPED_ONLY', 'id', 'id_mask', 'length', │ 'predicted.celltype.l2.score', 'age', │ ║ ║ │ 'quality', 'pos_hg19', 'id_hg19' │ 'organism_ontology_term_id', │ ║ ║ │ │ 'tissue_ontology_term_id', │ ║ ║ │ │ 'assay_ontology_term_id', │ ║ ║ │ │ 'disease_ontology_term_id', │ ║ ║ │ │ 'cell_type_ontology_term_id', │ ║ ║ │ │ 'self_reported_ethnicity_ontology_term_id', │ ║ ║ │ │ 'development_stage_ontology_term_id', │ ║ ║ │ │ 'sex_ontology_term_id', 'is_primary_data', │ ║ ║ │ │ 'suspension_type', 'tissue_type', 'cell_type', │ ║ ║ │ │ 'assay', 'disease', 'organism', 'sex', 'tissue', │ ║ ║ │ │ 'self_reported_ethnicity', 'development_stage', │ ║ ║ │ │ 'observation_joinid' │ ║ ║ │ uns: 'kinship' │ var: 'vst.mean', 'vst.variance', │ ║ ║ │ │ 'vst.variance.expected', │ ║ ║ │ │ 'vst.variance.standardized', 'vst.variable', │ ║ ║ │ │ 'feature_is_filtered', 'feature_name', │ ║ ║ │ │ 'feature_reference', 'feature_biotype', │ ║ ║ │ │ 'feature_length', 'feature_type', 'start', 'end', │ ║ ║ │ │ 'chrom' │ ║ ║ │ obsm: 'gPCs' │ uns: 'cell_type_ontology_term_id_colors', │ ║ ║ │ │ 'citation', 'default_embedding', │ ║ ║ │ │ 'schema_reference', 'schema_version', 'title' │ ║ ║ │ varm: 'filter' │ obsm: 'X_azimuth_spca', 'X_azimuth_umap', │ ║ ║ │ │ 'X_harmony', 'X_pca', 'X_umap' │ ║ ║ │ │ varm: 'PCs' │ ║ ║ └────────────────────────────────────────────────────┴────────────────────────────────────────────────────┘ ║ ╚═════════════════════════════════════════════════════════════════════════════════════════════════════════════╝
gdata = dd.G
gdata.var
| chrom | pos | a0 | a1 | AC | AC_Hemi | AC_Het | AC_Hom | AF | AN | ... | NS | R2 | TYPED | TYPED_ONLY | id | id_mask | length | quality | pos_hg19 | id_hg19 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| snp_id | |||||||||||||||||||||
| 2_563901_C_T | 2 | 563901 | C | T | 375 | 0 | 315 | 60 | 0.170765 | 2196 | ... | 1098 | 0.97602 | False | False | chr2_563901_C_T | False | 1 | NaN | 563901 | 2_563901_C_T |
| 2_841141_C_T | 2 | 841141 | C | T | 415 | 0 | 355 | 60 | 0.188980 | 2196 | ... | 1098 | 0.91015 | False | False | chr2_841141_C_T | False | 1 | NaN | 845199 | 2_845199_C_T |
| 2_1783323_C_A | 2 | 1783323 | C | A | 126 | 0 | 122 | 4 | 0.057377 | 2196 | ... | 1098 | 0.96989 | False | False | chr2_1783323_C_A | False | 1 | NaN | 1779551 | 2_1779551_C_A |
| 2_1855823_G_GTC | 2 | 1855823 | G | GTC | 432 | 0 | 364 | 68 | 0.196721 | 2196 | ... | 1098 | 0.97195 | False | False | chr2_1855823_G_GTC | False | 1 | NaN | 1852051 | 2_1852051_G_GTC |
| 2_1980881_C_T | 2 | 1980881 | C | T | 365 | 0 | 313 | 52 | 0.166211 | 2196 | ... | 1098 | 0.95519 | False | False | chr2_1980881_C_T | False | 1 | NaN | 1977109 | 2_1977109_C_T |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 22_50796327_CCA_C | 22 | 50796327 | CCA | C | 106 | 0 | 98 | 8 | 0.048270 | 2196 | ... | 1098 | 0.72635 | False | False | chr22_50796327_CCA_C | False | 3 | NaN | 50357898 | 22_50357898_CCA_C |
| 22_50798021_A_G | 22 | 50798021 | A | G | 149 | 0 | 139 | 10 | 0.067851 | 2196 | ... | 1098 | 0.76468 | False | False | chr22_50798021_A_G | False | 1 | NaN | 50359592 | 22_50359592_A_G |
| 22_50798635_T_C | 22 | 50798635 | T | C | 431 | 0 | 349 | 82 | 0.196266 | 2196 | ... | 1098 | 0.41777 | False | False | chr22_50798635_T_C | False | 1 | NaN | 50360206 | 22_50360206_T_C |
| 22_50799821_A_C | 22 | 50799821 | A | C | 144 | 0 | 136 | 8 | 0.065574 | 2196 | ... | 1098 | 0.70017 | False | False | chr22_50799821_A_C | False | 1 | NaN | 50361392 | 22_50361392_A_C |
| 22_50802428_C_G | 22 | 50802428 | C | G | 26 | 0 | 26 | 0 | 0.011840 | 2196 | ... | 1098 | 0.45162 | False | False | chr22_50802428_C_G | False | 1 | NaN | 50363999 | 22_50363999_C_G |
146939 rows × 25 columns
# gdata = gdata[:, :1000] # subset to some variants to run the noteobook faster
Export Variants to VCF#
Before running any annotation tools, we must export our variants from gdata into a standard VCF format. This file will serve as the input to tools like VEP, SnpEff, or FAVOR.
cl.io.write_variants_to_vcf(gdata, out_file=variant_file)
Run Variant Annotation Tools#
We support three different annotation tools: SnpEff, FAVOR, and VEP. Each tool offers different capabilities and plugin support. Below, we demonstrate how to run each of them using the cellink API. You can choose one or run all depending on your workflow needs.
Annotate with SnpEff#
SnpEff is a fast and widely used tool for predicting the effects of variants on genes and transcripts. Please install it via conda.
snpeff_annotation_file = str(DATA / "variant_annotation/variants_snpeff_annotated.txt")
cl.tl.run_snpeff(input_vcf=variant_file, output=snpeff_annotation_file)
Annotate with FAVOR#
FAVOR is particularly useful for integrating functional variant annotations from large public databases.
favor_annotation_file = str(DATA / "variant_annotation/variants_favor_annotated.txt")
cl.tl.run_favor(
database_dir="~/cellink_sample_data/favor",
G=gdata,
output=favor_annotation_file,
config_file=str(CONFIGS / "favor.yaml"),
)
Annotate with VEP#
VEP (Variant Effect Predictor) is an Ensembl tool that supports rich annotation via plugins. Here we show how to run it online, though we recommend using it offline for speed and plugin support. For running VEP offline and with plugins install the cache as explained in https://www.ensembl.org/info/docs/tools/vep/script/vep_cache.html#pre and relevant plugins and use vep_config_offline.yaml
Install VEP via
conda install bioconda::ensembl-vep
! vep
#----------------------------------#
# ENSEMBL VARIANT EFFECT PREDICTOR #
#----------------------------------#
Versions:
ensembl : 112.e71cae3
ensembl-funcgen : 112.be19ffa
ensembl-io : 112.2851b6f
ensembl-variation : 112.4113356
ensembl-vep : 112.0
Help: dev@ensembl.org , helpdesk@ensembl.org
Twitter: @ensembl
http://www.ensembl.org/info/docs/tools/vep/script/index.html
Usage:
./vep [--cache|--offline|--database] [arguments]
Basic options
=============
--help Display this message and quit
-i | --input_file Input file
-o | --output_file Output file
--force_overwrite Force overwriting of output file
--species [species] Species to use [default: "human"]
--everything Shortcut switch to turn on commonly used options. See web
documentation for details [default: off]
--fork [num_forks] Use forking to improve script runtime
For full option documentation see:
http://www.ensembl.org/info/docs/tools/vep/script/vep_options.html
vep_annotation_file = DATA / "variant_annotation/variants_vep_annotated.txt"
cl.tl.run_vep(
config_file=str(CONFIGS / "vep.yaml"), input_vcf=variant_file, output=vep_annotation_file
) # writes vep_annotation_file
Integrate Annotations into gdata#
Once annotation files are generated, we parse and add them to the gdata object. Initially, these annotations are stored in .uns, because there can be multiple annotations per variant (e.g., across different transcripts).
cl.tl.add_vep_annos_to_gdata(vep_anno_file=vep_annotation_file, gdata=gdata, dummy_consequence=True)
gdata.uns["variant_annotation_vep"]
| Consequence_downstream_gene_variant | Consequence_intergenic_variant | Consequence_intron_variant | Consequence_mature_miRNA_variant | Consequence_non_coding_transcript_exon_variant | Consequence_non_coding_transcript_variant | Consequence_splice_polypyrimidine_tract_variant | Consequence_splice_region_variant | Consequence_upstream_gene_variant | CADD_PHRED | ... | SIFT | gnomADe_NFE_AF | CADD_RAW | ENSP | gnomADe_FIN_AF | gnomADe_AF | Protein_position | STRAND | DISTANCE | CANONICAL | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| snp_id | gene_id | transcript_id | |||||||||||||||||||||
| 22_16388891_G_A | ENSG00000230643 | ENST00000447704 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | 594.0 | YES |
| 22_16388968_C_T | ENSG00000230643 | ENST00000447704 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | 517.0 | YES |
| 22_16389525_A_G | ENSG00000230643 | ENST00000447704 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16390411_G_A | ENSG00000230643 | ENST00000447704 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | 809.0 | YES |
| 22_16391555_G_C | ENSG00000230643 | ENST00000447704 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | 1953.0 | YES |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 22_16929238_C_T | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16929483_A_G | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16929768_C_A | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16929834_A_C | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1.401 | ... | NaN | NaN | -0.100515 | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16929855_C_G | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0.453 | ... | NaN | NaN | -0.268616 | - | NaN | NaN | NaN | -1.0 | NaN | YES |
1077 rows × 39 columns
Combine Annotations Across Tools#
If you’ve annotated with multiple tools (e.g., VEP + FAVOR), you can combine their results into a unified table. This allows downstream functions to access a single, harmonized view of all annotations.
cl.tl.combine_annotations(gdata, ["vep"])
gdata
AnnData object with n_obs × n_vars = 981 × 1000
var: 'chrom', 'pos', 'a0', 'a1', 'AC', 'AC_Hemi', 'AC_Het', 'AC_Hom', 'AF', 'AN', 'ER2', 'ExcHet', 'HWE', 'IMPUTED', 'maf', 'NS', 'R2', 'TYPED', 'TYPED_ONLY', 'id', 'id_mask', 'length', 'quality', 'pos_hg19', 'id_hg19'
uns: 'kinship', 'variant_annotation_vep', 'variant_annotation'
obsm: 'gPCs'
varm: 'filter'
gdata.uns["variant_annotation"]
| Consequence_downstream_gene_variant | Consequence_intergenic_variant | Consequence_intron_variant | Consequence_mature_miRNA_variant | Consequence_non_coding_transcript_exon_variant | Consequence_non_coding_transcript_variant | Consequence_splice_polypyrimidine_tract_variant | Consequence_splice_region_variant | Consequence_upstream_gene_variant | CADD_PHRED | ... | SIFT | gnomADe_NFE_AF | CADD_RAW | ENSP | gnomADe_FIN_AF | gnomADe_AF | Protein_position | STRAND | DISTANCE | CANONICAL | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| snp_id | gene_id | transcript_id | |||||||||||||||||||||
| 22_16388891_G_A | ENSG00000230643 | ENST00000447704 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | 594.0 | YES |
| 22_16388968_C_T | ENSG00000230643 | ENST00000447704 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | 517.0 | YES |
| 22_16389525_A_G | ENSG00000230643 | ENST00000447704 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16390411_G_A | ENSG00000230643 | ENST00000447704 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | 809.0 | YES |
| 22_16391555_G_C | ENSG00000230643 | ENST00000447704 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | 1953.0 | YES |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 22_16929238_C_T | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16929483_A_G | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16929768_C_A | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | NaN | ... | NaN | NaN | NaN | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16929834_A_C | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1.401 | ... | NaN | NaN | -0.100515 | - | NaN | NaN | NaN | -1.0 | NaN | YES |
| 22_16929855_C_G | ENSG00000227367 | ENST00000421957 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0.453 | ... | NaN | NaN | -0.268616 | - | NaN | NaN | NaN | -1.0 | NaN | YES |
1077 rows × 39 columns
Aggregate Context-Specific Annotations#
Each variant may map to multiple genes or transcripts, resulting in multiple annotation rows per variant. This is why the variant annotations are initially stored in uns. To be able to set the annotations as varm and to simplify downstream use (e.g., filtering or visualization), we aggregate these into a single row per variant using strategies such as:
first: Keep the first unique value.unique_list_max: Merge strings as comma-separated lists and take max for numeric fields.
This allows us to move annotations from .uns to .varm for better integration with the AnnData structure.
print(len(gdata.uns["variant_annotation_vep"].columns))
for agg_type in ["first", "unique_list_max"]: # "list", "str"]:
res = cl.tl.aggregate_annotations_for_varm(gdata, "variant_annotation_vep", agg_type=agg_type, return_data=True)
print(len(res))
print(len(res.columns))
gdata
39
1000
41
1000
41
AnnData object with n_obs × n_vars = 981 × 1000
var: 'chrom', 'pos', 'a0', 'a1', 'AC', 'AC_Hemi', 'AC_Het', 'AC_Hom', 'AF', 'AN', 'ER2', 'ExcHet', 'HWE', 'IMPUTED', 'maf', 'NS', 'R2', 'TYPED', 'TYPED_ONLY', 'id', 'id_mask', 'length', 'quality', 'pos_hg19', 'id_hg19'
uns: 'kinship', 'variant_annotation_vep', 'variant_annotation'
obsm: 'gPCs'
varm: 'filter', 'variant_annotation'
cl.tl.aggregate_annotations_for_varm(gdata, "variant_annotation_vep", agg_type="unique_list_max", return_data=True)
| Amino_acids | BIOTYPE | CADD_PHRED | CADD_RAW | CANONICAL | CDS_position | CLIN_SIG | Codons | Consequence_downstream_gene_variant | Consequence_intergenic_variant | ... | gnomADe_AF | gnomADe_AFR_AF | gnomADe_AMR_AF | gnomADe_ASJ_AF | gnomADe_EAS_AF | gnomADe_FIN_AF | gnomADe_NFE_AF | gnomADe_OTH_AF | gnomADe_SAS_AF | transcript_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| snp_id | |||||||||||||||||||||
| 22_16388891_G_A | NaN | unprocessed_pseudogene | NaN | NaN | YES | NaN | NaN | NaN | 1 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000447704 |
| 22_16388968_C_T | NaN | unprocessed_pseudogene | NaN | NaN | YES | NaN | NaN | NaN | 1 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000447704 |
| 22_16389525_A_G | NaN | unprocessed_pseudogene | NaN | NaN | YES | NaN | NaN | NaN | 0 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000447704 |
| 22_16390411_G_A | NaN | unprocessed_pseudogene | NaN | NaN | YES | NaN | NaN | NaN | 0 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000447704 |
| 22_16391555_G_C | NaN | unprocessed_pseudogene | NaN | NaN | YES | NaN | NaN | NaN | 0 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000447704 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 22_16929238_C_T | NaN | unprocessed_pseudogene | NaN | NaN | YES | NaN | NaN | NaN | 0 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000421957 |
| 22_16929483_A_G | NaN | unprocessed_pseudogene | NaN | NaN | YES | NaN | NaN | NaN | 0 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000421957 |
| 22_16929768_C_A | NaN | unprocessed_pseudogene | NaN | NaN | YES | NaN | NaN | NaN | 0 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000421957 |
| 22_16929834_A_C | NaN | unprocessed_pseudogene | 1.401 | -0.100515 | YES | NaN | NaN | NaN | 0 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000421957 |
| 22_16929855_C_G | NaN | unprocessed_pseudogene | 0.453 | -0.268616 | YES | NaN | NaN | NaN | 0 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000421957 |
1000 rows × 41 columns
gdata.varm["variant_annotation"].query('BIOTYPE == "protein_coding"')
| Amino_acids | BIOTYPE | CADD_PHRED | CADD_RAW | CANONICAL | CDS_position | CLIN_SIG | Codons | Consequence_downstream_gene_variant | Consequence_intergenic_variant | ... | gnomADe_AF | gnomADe_AFR_AF | gnomADe_AMR_AF | gnomADe_ASJ_AF | gnomADe_EAS_AF | gnomADe_FIN_AF | gnomADe_NFE_AF | gnomADe_OTH_AF | gnomADe_SAS_AF | transcript_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| snp_id | |||||||||||||||||||||
| 22_16451723_A_T | NaN | protein_coding | NaN | NaN | YES | NaN | NaN | NaN | 0 | 0 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | ENST00000252835 |
1 rows × 41 columns
Test writing#
gdata.uns.pop("variant_annotation_vep", None) # hast to be dropped because of multiindex before writing
gdata.uns.pop("variant_annotation", None) # hast to be dropped because of multiindex before writing
gdata.write("gdata.h5ad") # test
Explore and Visualize Variant Annotations#
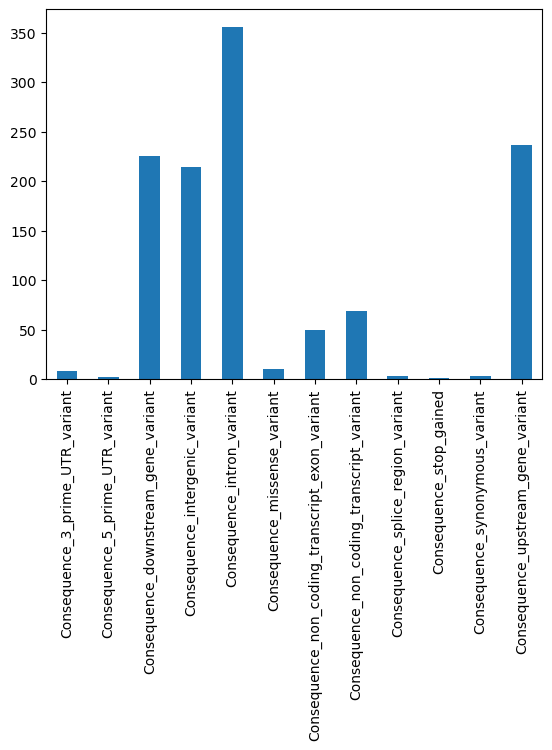
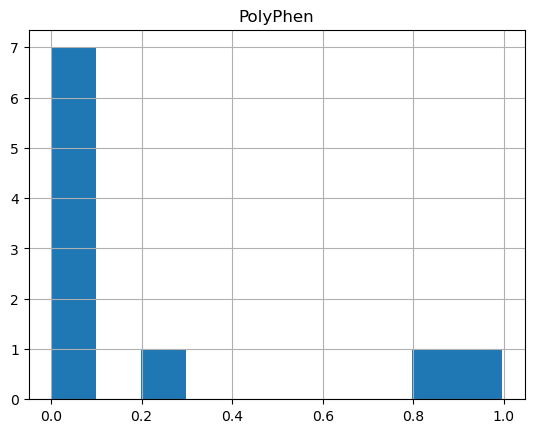
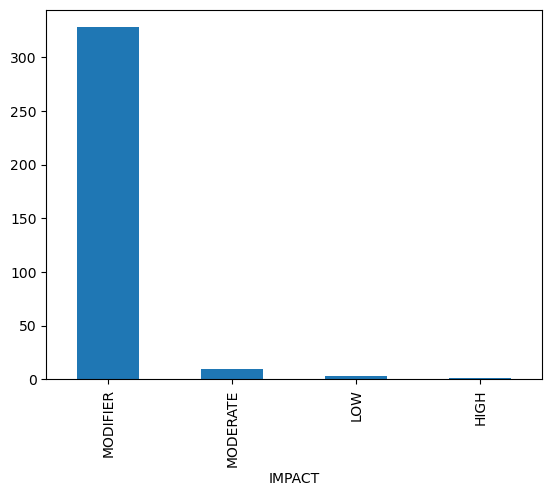
We now explore the aggregated annotations stored in gdata.varm["variant_annotation"]. We’ll focus on protein-coding variants and visualize:
The distribution of predicted impact categories.
PolyPhen scores, which predict damaging effects of coding variants.
Frequencies of different consequence types.
These plots help assess the functional relevance of the annotated variants.
gdata.varm["variant_annotation"].columns
Index(['Amino_acids', 'BIOTYPE', 'CANONICAL', 'CDS_position', 'Codons',
'Consequence_3_prime_UTR_variant', 'Consequence_5_prime_UTR_variant',
'Consequence_downstream_gene_variant', 'Consequence_intergenic_variant',
'Consequence_intron_variant', 'Consequence_missense_variant',
'Consequence_non_coding_transcript_exon_variant',
'Consequence_non_coding_transcript_variant',
'Consequence_splice_region_variant', 'Consequence_stop_gained',
'Consequence_synonymous_variant', 'Consequence_upstream_gene_variant',
'DISTANCE', 'ENSP', 'Existing_variation', 'FLAGS', 'IMPACT', 'PolyPhen',
'Protein_position', 'SIFT', 'STRAND', 'cDNA_position', 'gene_id',
'transcript_id'],
dtype='object')
gdata.varm["variant_annotation"].query('BIOTYPE == "protein_coding"')[["PolyPhen"]].hist()
array([[<Axes: title={'center': 'PolyPhen'}>]], dtype=object)
gdata.varm["variant_annotation"].query('BIOTYPE == "protein_coding"')["IMPACT"].value_counts().plot(kind="bar")
<Axes: xlabel='IMPACT'>
consequence_cols = [col for col in gdata.varm["variant_annotation"].columns if "Consequence" in col]
gdata.varm["variant_annotation"][consequence_cols].sum().plot(kind="bar")
<Axes: >